![]()
Smith-Kingsmore Syndrome Foundation
Whether you’re a parent or family member seeking more information to help your child with Smith-Kingsmore syndrome (SKS), a supportive friend, a doctor, or a researcher, you’ve come to the right place. We hope you find the information here useful, and will join us in investing in SKS research by donating now. Welcome! We’re glad you found us.
 SKS Foundation is proud to be part of the Chan Zuckerberg Initiative #RareAsOne Network – a group of 50 patient-led organizations that are strengthening rare disease communities, improving diagnosis, accelerating research and driving progress in the fight against rare diseases.
SKS Foundation is proud to be part of the Chan Zuckerberg Initiative #RareAsOne Network – a group of 50 patient-led organizations that are strengthening rare disease communities, improving diagnosis, accelerating research and driving progress in the fight against rare diseases.

Guide for Families
Click here for a copy of the SKS Family Guide, available in multiple languages, to share information on SKS with friends, family and medical professionals.
Some Science
Smith-Kingsmore syndrome (SKS) is a rare, neurodevelopmental genetic disorder caused by changes (disease-causing variants) in the MTOR gene. SKS impacts the digestive, endocrine, metabolic and nervous systems.
Patients with SKS have a spectrum of medical, intellectual, and behavioral disabilities resulting in different and variable clinical outcomes. The most common findings in people with SKS are intellectual disability, developmental delay, large brain size (megalencephaly) and seizures[1]. The symptoms vary and largely depend on the type of MTOR gene mutation that the patient has and its distribution in the body. Management of patients with SKS includes treatment of medical concerns, such as seizures as well as speech and physical therapy. Treatment is symptom-driven and there is currently no cure.
Changes in MTOR Gene
Genetic changes in the MTOR gene were first noted as a cause of a neurodevelopmental disorder in 2013[2]. Studies are still needed to continue to define the characteristics associated with specific MTOR gene variants. Presently, genetic changes in MTOR can be separated into three clinical types[3]:
- The first group includes patients with generalized brain overgrowth (megalencephaly), intellectual disability, autism and hypotonia. These are the patients who have been identified as having SKS.
- The second group includes patients with diffuse brain overgrowth, abnormalities of the surface of the brain (polymicrogyria) and skin pigmentation abnormalities.
- The third group includes patients with focal changes in the brain (focal cortical dysplasia or hemimegalencephaly) causing early-onset epilepsy.
The distribution and levels of MTOR genetic changes in these three groups vary. The mutations in MTOR in patients with SKS are usually present in all cells of the body. Disease characteristics vary in patients with SKS, even if they have the same MTOR gene mutation. In contrast, genetic changes in MTOR in the second and third groups can be more localized or restricted to certain tissues (mosaic). This may cause these mutations to escape detection if a patient has genetic testing on a blood or saliva sample.
Causes of Smith-Kingsmore Syndrome
Smith-Kingsmore syndrome is usually an autosomal dominant condition, which means that one copy of the altered MTOR gene in each cell is sufficient to cause the disorder.
Changes in the MTOR gene are usually random events (sporadic or de novo) that happen in the egg or sperm prior to conception and are not inherited from either parent. This type of change is present in all cells of the affected individual and is called a germline variant.
There are also some SKS patients who have an altered MTOR gene in some, but not all of their cells, and this is called somatic mosaicism. This type of change is also de novo (not inherited) and occurs at some point while a baby is developing during pregnancy. MTOR gene mutations in these SKS patients can only be detected in samples of affected tissues and might not be detected in a blood or saliva sample.
Rarely, people with SKS inherit the altered gene from an unaffected parent who has a MTOR gene mutation only in their sperm or egg cells (germline tissues). This is called germline mosaicism and, although rare, it has been seen more frequently in SKS than in other diseases.
MTOR Gene and the MTOR Pathway
The MTOR gene encodes the MTOR protein which plays a central role in the cell’s nutrient/energy sensing pathway. This pathway provides a way for cells to communicate information such as when to grow and how quickly to grow. Changes in the MTOR gene may change the instructions for the body and how the MTOR pathway works.
Changes in MTOR can cause the pathway to become hyperactive (i.e., gain of function). As a result of pathway hyperactivation, the affected nerve cells (neurons) grow unusually large and misshapen, leading to brain malformations, cognitive delays and epilepsy.
The MTOR protein is a core component of two protein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).

Gordo G, Tenorio J, Arias P, et al (2018)[1]
In addition to Smith-Kingsmore syndrome, mutations in different genes in this pathway result in many other neurodevelopmental disorders. These include PIK3CA related overgrowth disorders, Tuberous Sclerosis, PTEN-related disorders (including the macrocephaly-autism syndrome), among many others, with possible clinical overlap between these disorders and MTOR related disorders.
SKS Variants
To date the majority of disease-causing mutations identified in MTOR in patients with Smith-Kingsmore syndrome are missense changes. Mutations in the focal adhesion targeting (FAT) domain are frequent, especially the recurrent variant p.Glu1799Lys. Research has also found changes in the Kinase and other domains, including: p.Phe594Ser, p.Gln1014His, p.Lys1395Arg, p.Arg1480_Cys1483del, p.Arg1482Cys, p.Arg1482Pro, p.Cys1483Phe, p.Cys1483Tyr, p.Trp1490Arg, p.Ala1519Thr, p.Met1595Ile, p.Glu1799Lys, p.Ala1832Thr, p.Phe1888Cys, p.Phe1888Ser, p.Ala1971Thr, p.Thr1977Ile, p.Val2006Leu, p.Arg2110Gln, p.Phe2202Cys, p.Ser2215Phe, p.Met2327Ile, p.Gly2359Glu, p.Val2406Met, p.Gly2464Val, p.Ile2501Val.
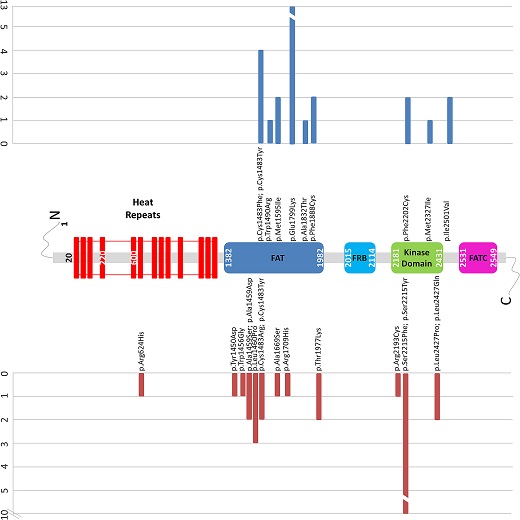
Gordo G, Tenorio J, Arias P, et al (2018)[1]
Affected Populations
MTOR-related disorders are extremely rare. The total patient population is still unknown, but it is estimated that there are about 10 people with MTOR gene disorders (with some MTOR mutations causing SKS) in every 10,000 individuals. However, SKS may go undiagnosed or misdiagnosed, making it extremely difficult to determine the true frequency in the general population. Based on the current understanding of this condition, SKS occurs worldwide in people of all ethnic groups.
Standard Therapies
Currently, there is no cure for Smith-Kingsmore syndrome, and no treatments approved by the U.S. Food and Drug Administrations (FDA). Treatment is based on a child’s specific symptoms. Patients and their families typically visit one or more of the following medical specialists:
Pediatrics
- Annual visits to monitor growth and development
- Medical management of constipation is often needed
- Monitoring for illness due to abnormal immune cell function
Developmental Pediatrics
- Developmental and behavioral evaluations to assess for challenges and recommend treatments
- Evaluate for appropriate therapies including physical, occupational, speech/feeding, behavioral, vision therapy
- Guide individualized education plans (IEPs)
Pediatric Genetics and Genetic Counseling
- Review genetic testing and results
- Provide information about recurrence risk
- Provide coordination of care
Neurology
- If seizures are suspected, an EEG (measurement of the brain’s electrical activity) is recommended
- An MRI should be considered to identify any brain malformations
Ophthalmologist/Neuro-ophthalmologist
- Screening for cortical visual impairment (CVI)
Audiology
- Routine hearing screening (newborn and annually)
Endocrinology
- Consider a referral if hypoglycemia develops or if premature (precocious) puberty is suspected Orthopedics/Physical Rehabilitation
- Evaluate the need for assistive devices due to hypotonia, motor deficits and/or bone abnormalities
Neuropsychology
- For school-age children, this assessment can help identify the most appropriate educational support and schooling
Routine dental and/or orthodontic care is also recommended as well as speech and language therapy, physical and occupational therapy and behavior therapy/psychological counseling.
Investigational Therapies
Precision is particularly important for MTOR. As the master energy sensor, it regulates metabolic homeostasis in each cell. Too much or too little activity can have adverse effects in cells and the whole-body physiology. Most people with SKS have higher protein activity levels, but not equally higher than normal. Our goal is to find precision and personalized treatment options to bring cellular activity into balance. Enhancing cellular homeostasis will improve lives of people with SKS, and possibly prevent future devastating diseases, such as cancer.
Researchers continue to learn more about Smith-Kingsmore syndrome and other conditions caused by MTOR mutations. By better understanding the characteristics and the differences of MTOR conditions like SKS, they can design well-informed, precision treatment plans. Some patients with SKS have been prescribed sirolimus (rapamycin) or everolimus to treat intractable seizures (seizures that cannot be controlled completely by other medications). There is currently no published data about how well this works (efficacy) and these drugs are not currently approved by the FDA to treat SKS. Studies are pending to determine the long-term effects of rapamycin on neurocognitive development in people with SKS and clinical trials are needed to clarify potential effectiveness of rapamycin.
Other Names
- SKS
- Macrocephaly-Intellectual disability-Neurodevelopmental Disorder-Small thorax syndrome
- MINDS syndrome





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}